





APA
ISO 690-2
Harvard
Haga clic en un formato de citación
Fenotipo NMOSD-like recurrente en un paciente pediátrico con enfermedad por anticuerpos anti-MOG: presentación de caso
Recurrent NMOSD-like Phenotype in a Pediatric Patient with Myelin Oligodendrocyte Glycoprotein Antibody Disease: A Case Report
Isabella Lince-Rivera ![]() , Natalia Martínez-Córdoba
, Natalia Martínez-Córdoba ![]() , Andrés Felipe Araújo Polanía
, Andrés Felipe Araújo Polanía ![]()
Fenotipo NMOSD-like recurrente en un paciente pediátrico con enfermedad por anticuerpos anti-MOG: presentación de caso
Salud Javeriana, vol. 2, 2025
Pontificia Universidad Javeriana
Isabella Lince-Rivera a est.isabella.lince@unimilitar.edu.co
Pontificia Universidad Javeriana, Colombia
Natalia Martínez-Córdoba
Universidad Militar Nueva Granada, Bogotá, Colombia
Andrés Felipe Araújo Polanía
Hospital Militar Central, Bogotá, Colombia
Recibido: 21 diciembre 2024
Aceptado: 11 abril 2025
Resumen: Introducción: La enfermedad por anticuerpos anti-MOG (MOGAD) tiene una presentación clínica variable. El fenotipo ADEM (encefalomielitis aguda diseminada) es el más frecuente en los pacientes pediátricos más pequeños, y en pacientes mayores, la neuritis óptica y la mielitis transversa. El fenotipo NMOSD-like incluye a aquellos pacientes con episodios de neuritis ópticao mielitis transversa con síndromes de tallo o sin estos. Presentación del caso: Niño de 11 años, quien presentó un cuadro de neuritis óptica, seguido de dos síndromes de tallo con afectación cerebelosa, con resultado de anticuerpos (Ac) antiacuaporina 4 (AQP4) en suero y bandas oligoclonales en el líquido cefalorraquídeo negativos, y anticuerpos anti-MOG en suero positivos. Recibió manejo en la etapa aguda con corticoesteroide endovenoso y tuvo una adecuada respuesta. Conclusiones: Se debe sospechar MOGAD en pacientes pediátricos con síndromes desmielinizantes y solicitar anticuerpos anti-MOG, especialmente en pacientes con Ac anti-AQP4 y bandas oligoclonales negativos. El diagnóstico temprano y el tratamiento modificador de la enfermedad es de gran importancia para prevenir recurrencias, evitar secuelas y mejorar la calidad de vida de estos pacientes.
Palabras clave:anti-MOG, desmielinización, neuritis óptica, MOGAD, pediatría.
Abstract: Introduction: Myelin Oligodendrocyte Glycoprotein antibody disease (MOGAD) has a variable clinical presentation, with ADEM being the most frequent phenotype in younger pediatric patients and optic neuritis and transverse myelitis in older patients. The NMOSD-like phenotype includes episodes of optic neuritis and/or transverse myelitis with or without brainstem syndromes. Case presentation: An 11-year-old male patient presented with symptoms of optic neuritis, followed by two brainstem syndromes with cerebellar involvement, and negative aquaporin 4 antibodies (AQP4) and oligoclonal bands, with positive MOG antibodies. He was treated in the acute stage with intravenous corticosteroid with adequate response. Conclusions:MOGAD should be suspected in pediatric patients with demyelinating syndromes, and MOG antibodies should be requested, especially in patients with negative AQP4 antibodies and oligoclonal bands. Early diagnosis and maintenance therapy is important to prevent recurrences, avoid sequelae, and improve the quality of life of these patients.
Keywords: anti-MOG, demyelination, MOGAD, optic neuritis, pediatrics.
Introducción
La glucoproteína de la mielina del oligodendrocito (MOG, por sus siglas en inglés) es una de las proteínas producidas por los oligodendrocitos, que son las células que generan la mielina en el sistema nervioso central (SNC) (1). Así como la proteína básica de la mielina (MBP, por sus siglas en inglés), la proteína proteolipídica y la glucoproteína asociada a la mielina, la MOG es un componente esencial de la mielina, que está expresado en el SNC (1). Se localiza en la superficie externa de la vaina de la mielina y en la membrana plasmática de los oligodendrocitos (2,3). Dentro de sus funciones está la formación y mantenimiento de la integridad estructural de la vaina de la mielina, la comunicación intercelular, la mediación de interacciones entre la mielina y el sistema inmune, y la regulación de la estabilidad de los microtúbulos en los oligodendrocitos (1,4). Dada su localización y estructura, con dominio extracelular, es un blanco fácilmente accesible para los autoanticuerpos (figura 1) (1,5).
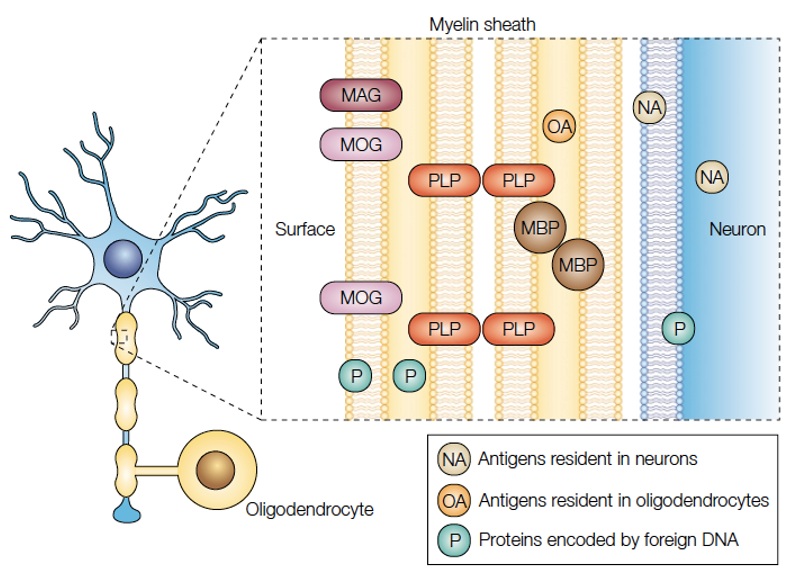
La expresión de la MOG empieza cuando inicia la mielinización. De hecho, se ha descrito como un posible marcador de maduración de los oligodendrocitos, y se cree que el grado de desarrollo de la mielina y su compactación —lo cual se relaciona directamente con la edad— tienen un rol importante en el fenotipo clínico que presentan los pacientes con anticuerpos (Ac) anti-MOG positivos. Por esta razón, se habla de fenotipos dependientes de la edad y se ha descrito que la encefalomielitis aguda diseminada (ADEM) es más frecuente en los pacientes menores; mientras que el síndrome óptico-espinal lo es en pacientes mayores (4,7). También en niños menores se han descrito cuadros clínicos más severos, pero con una recuperación más rápida y completa, y con una menor tasa de recurrencia (3,8).
En términos de fisiopatología, se desconoce el desencadenante claro para la producción de Ac anti-MOG, pero se cree que la inducción autoinmune ocurre en el sistema inmune periférico (8). Aunque la autoinmunidad posinfecciosa es un desencadenante probable, no se han identificado patógenos específicos de la enfermedad. Se cree que existe una disrupción de las barreras del SNC, que permite el paso de células B, células plasmáticas y autoanticuerpos y permiten su interacción con el autoantígeno (MOG) expresado en la mielina, que la daña y lleva a una desmielinización subsecuente. En paralelo, los Ac anti-MOG y las células plasmáticas también pueden estimular la activación de células T CD4 específicas para MOG, células T efectoras específicas de proteína básica de la mielina y macrófagos del SNC.
En relación con lo anterior, usualmente aumentan las citocinas proinflamatorias en el líquido cefalorraquídeo (LCR), como la interleucina 6 (IL-6), la IL-17, el factor estimulador de colonias de granulocitos, el factor de necrosis tumoral α, y las quimioquinas, por ejemplo, el factor activador de células B, el ligando inductor de proliferación, la quimioquina ligando 13 y la quimoquina ligando 19 (8).
La presentación clínica de la enfermedad es variable. La ADEM es la forma más frecuente de la MOGAD pediátrica y ocurre predominantemente en la infancia temprana, tras una infección o una vacunación (3,5). Se ha evidenciado que hasta el 50 %-60 % de niños con ADEM y casi todos los niños que recurren tras la ADEM (encefalitis diseminada multifásica y ADEM con posterior neuritis óptica) tienen títulos altos de Ac anti-MOG (7). Pacientes mayores presentan con mayor frecuencia neuritis óptica, mielitis transvers y existe en ellos un fenotipo NMOSD-like (7).
Existen fenotipos clínicos emergentes relacionados con la MOGAD, como la encefalitis cortical cerebral, el síndrome de tallo, el ataque desmielinizante cerebeloso, las neuropatías craneales, el fenotipo leucodistrofia-like (que se ha categorizado como un fenotipo de mal pronóstico), la desmielinización combinada (central y periférica) y los casos no clasificables (que son aquellos que no cumplen los criterios para ningún fenotipo relacionado con MOGAD hasta el momento) (5,8).
Usualmente, los pacientes presentan estos fenotipos de forma aislada, pero en ocasiones o bien aparecen combinados, o bien suelen ser recurrentes. Esto último se define como un nuevo evento clínico que ocurre más de 30 días tras el inicio de un evento previo, y en MOGAD aparece con mayor frecuencia en los primeros 2 años tras el primer evento (1,9).
Respecto a los fenotipos clínicos recurrentes, pacientes mayores, que permanecen seropositivos en el seguimiento, que cursan inicialmente con neuritis óptica y en los que pasa una menor cantidad de tiempo entre el primer evento y la primera recurrencia, tienen mayor riesgo de recurrir (10,11). Algunos estudios han mostrado que los niveles de anticuerpos parecen no tener relación con el riesgo de recurrencia (11). En menores de 9 años, lo más frecuente es la encefalomielitis diseminada multifásica y la ADEM seguida de neuritis óptica; entre tanto, en pacientes mayores de 9 años, es la neuritis óptica recurrente y el fenotipo NMOSD-like recurrente (5).
Antes, en sospecha de MOGAD, se recomendaba el uso de ELISA, inmunohistoquímica o Western blot como técnicas de laboratorio para identificar los anticuerpos anti-MOG; sin embargo, actualmente no deben usarse (1), dado que nuevas técnicas como ensayos basados en células han mostrado mayor especificidad, especialmente cuando se realiza en suero (4). Los niveles de inmunoglobulina G (IgG), considerados significativos por este método, son ≥1:100 (9). Otros estudios paraclínicos, como la punción lumbar con medición del recuento celular, proteínas, índice de IgG y bandas oligoclonales, y las neuroimágenes (según sus hallazgos y progresión de lesiones de forma asintomática) son útiles para diferenciar MOGAD de otros diagnósticos que se presentan con los mismos fenotipos clínicos, como la esclerosis múltiple (EM) y los trastornos del espectro de la neuromielitis óptica (NMOSD) (1).
Según el panel internacional de MOGAD, para diagnosticar esta patología, se deben cumplir los siguientes tres criterios: a) un evento clínico desmielinizante típico, dentro de los cuales se incluyen neuritis óptica, mielitis, ADEM, déficits cerebrales mono o polifocales, déficits de tallo o cerebelosos y encefalitis cortical cerebral frecuentemente asociada a crisis epilépticas; b) anticuerpos IgG anti-MOG positivos (en suero por ensayo basado en células), y c) exclusión de otros diagnósticos como EM (9). Es relevante tener en cuenta que, respecto al criterio b, si se tiene un reporte claramente positivo de anticuerpos, no se requieren hallazgos adicionales; sin embargo, en caso de que sea un reporte positivo bajo, positivo sin títulos reportados o negativo en suero pero positivo en LCR, se deben tener anticuerpos AQP4 negativos y uno o más hallazgos en la resonancia magnética nuclear (RMN) sugestivos de MOGAD, según el fenotipo para cumplir este criterio.
No existen guías basadas en la evidencia para el tratamiento agudo y de mantenimiento en MOGAD (12); pero en 2020 se publicaron las recomendaciones del Consenso Europeo. En el contexto agudo, se recomienda recopilar todos los hallazgos clínicos e imagenológicos, y si según estos el caso es altamente sugestivo de MOGAD, se debe manejar con metilprednisolona endovenosa a 20-30 mg/kg cada día (máximo 1 g/día) durante 3-5 días (8,13). Varios estudios han demostrado que los pacientes con MOGAD son altamente sensibles a los corticoesteroides, y en muchos casos logran una remisión completa de los síntomas tras este ciclo corto endovenoso (1). No obstante, en caso de no obtener respuesta o tener una respuesta insuficiente, se recomienda el uso de inmunoglobulina endovenosa con un total de 1-2 g/kg en 1-5 días sin exceder 1 g/kg/día, con la mayoría de expertos recomendando una duración de 5 días (8,13). En episodios severos se recomiendan hasta 5 ciclos de plasmaféresis.
Se ha discutido uso de corticoesteroides orales después del primer evento desmielinizante, principalmente si el paciente persiste con síntomas luego del ciclo endovenoso, con una vigilancia estricta durante su descenso y suspensión, dado el riesgo de recurrencia (13).
A continuación se presenta el caso de un paciente adolescente con enfermedad por anticuerpos anti-MOG con un fenotipo NMOSD-like recurrente, un síndrome raro para su edad. En él se documentó un realce leptomeníngeo asociado con pleocitosis previo a su última recaída. Ello apoyaría estudios que han planteado estos hallazgos como marcadores de riesgo de recurrencia (3,14). Se contó con una aprobación escrita de la institución médica (código 92023) y con el consentimiento informado y firmado por el padre del paciente. Los procedimientos para su reporte siguieron las normas éticas establecidas concordantes con la Declaración de Helsinki de la Asociación Médica Mundial.
Presentación del caso clínico
El caso corresponde a un preadolescente de 11 años de edad sin historia de cefalea, quien ingresó a urgencias de pediatría por un cuadro clínico de 15 días de evolución, consistente en dolor holocraneano de predominio frontal, opresivo, de intensidad y frecuencia progresivas, que lo despertaba en las noches, sin respuesta al manejo analgésico, asociado con fotofobia, dolor lumbar y parestesias en sus miembros inferiores en los últimos 7 días. Adicionalmente, en los últimos 5 días del cuadro describía sensación de debilidad generalizada, dolor en los miembros inferiores, sensación vertiginosa, dificultad para la marcha y aumento del polígono de sustentación. Negó asociación con algún desencadenante, negó síntomas constitucionales e infecciosos en el momento, y refirió un cuadro viral hacía un mes.
En el examen físico se identificó dolor a la palpación de la región dorsolumbar medial, aparente debilidad en los miembros inferiores, marcha con aumento del polígono de sustentación y dificultad para incorporarse; sin embargo, el paciente era poco colaborador. Se consideró hospitalizar y realizarle una RMN de neuroeje. También se solicitó electromiografía y neuroconducciones (EMG + NCS) de las cuatro extremidades y exámenes paraclínicos séricos, incluidas creatina fosfocinasa y transaminasas —cuyo resultado fue normal—, y se inició manejo analgésico.
Al día siguiente, inició con disminución de la agudeza visual progresiva: primero en el ojo derecho y después en ambos ojos. El resultado de la RMN cerebral simple mostró una lesión hiperintensa única redondeada de contornos bien definidos, discretamente hiperintensa en T1, con prolongación de la difusión en la sustancia blanca del centro semioval izquierdo, de naturaleza inespecífica (figura 2). La RMN de columna no evidenció alteraciones. Impresionó también una hiperseñal de los nervios ópticos.

Dados los hallazgos clínicos y paraclínicos, se solicitó que el servicio de oftalmología valorara al paciente. Allí se identificaron dolor con los movimientos oculares y papiledema de ambos ojos. Se le realizó una punción lumbar con presión de apertura de 10,5 cm H.O, aspecto transparente, color agua de roca, glucosa: 54 mg/dl, glucosa central: 99 mg/dl, hematíes de 250 y 110 leucocitos con 80 % de linfocitos. Proteínas totales: 65,4 mg/dl, y tinta china, Gram y cultivo negativos. Se consideró pleocitosis con predominio de linfocitos y leve elevación de proteínas, sin hipoglucorraquia en relación con un posible proceso inflamatorio o infeccioso. Se solicitaron los exámenes FilmArray, prueba de adenosina desaminasa y reacción en cadena de la polimerasa para tuberculosis, que resultaron negativos, y perfil autoinmune, que resultó normal.
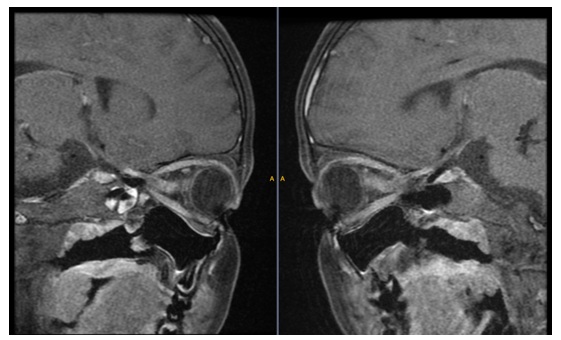
Continuó con deterioro de la agudeza visual llegando a visión en cuenta dedos y persistió el dolor ocular intermitente. Se solicitaron potenciales visuales que resultaron con periodos de latencia prolongados y RMN contrastada de órbitas, en la que se evidenció realce de los nervios ópticos con el contraste (figura 3). Se consideró que el paciente tenía una neuritis óptica y se inició manejo con corticoide: metilprednisolona 1 g/día durante 3 días. Así, mejoró de manera significativa la agudeza visual y el edema de papila bilateral. Se consideró continuar con prednisolona a 1 mg/kg cada día durante 2 semanas y se dio egreso.
Dos meses después, el paciente reingresó por cuadro clínico de 8 días de evolución de síntomas respiratorios (rinorrea hialina y tos húmeda), cefalea de predominio frontal de intensidad leve a moderada, visión doble y sensación vertiginosa. Durante la evaluación, se evidenció Romberg positivo, aumento del polígono de sustentación, dismetría y diplopía binocular horizontal. Se solicitó una RMN cerebral simple y contrastada y valoración por otorrinolaringología y oftalmología.
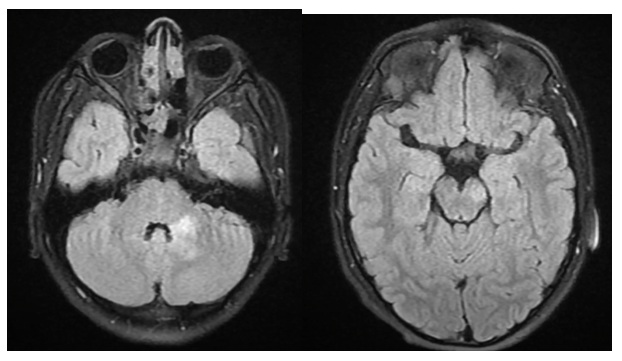
El servicio de otorrinolaringología realizó pruebas vestibulares y consideró que el paciente padecía vértigo de origen central; entre tanto, el servicio de oftalmología evidenció paresia del VI par izquierdo, e indicó oclusión ocular. En cuanto al reporte de RMN cerebral, se registraron hiperseñales irregulares mal definidas en el pedúnculo cerebeloso medio del lado izquierdo, así como en el pie del pedúnculo cerebral del mismo lado, con realce de manera difusa heterogénea lineal e irregular con el medio de contraste, sin modificarse en las secuencias de la difusión, de naturaleza más probablemente desmielinizante (figura 4). La RMN de columna fue normal.
Se consideró que el paciente estaba cursando con un segundo evento desmielinizante; un síndrome de tallo con compromiso cerebeloso y sospecha de EM. Una nueva punción lumbar reveló células 0, proteínas de 8 mg/dl, glucosa de 60 mg/dl, bandas oligoclonales que no se pudieron procesar por un error de laboratorio, y Ac anti-AQ4 en suero negativos. Se indicó un nuevo ciclo de corticoide endovenoso, con mejoría de síntomas, y por los dos eventos desmielinizantes descritos se le diagnosticó al paciente EM de inicio en la infancia, según los criterios de McDonald, y se decidió iniciar acetato de glatiramer. Durante los controles ambulatorios, el paciente permaneció en manejo sin recaídas, sin síntomas y con dos RMN cerebrales sin evidencia de lesiones, por lo que se consideró un curso clínico poco probable de EM y se decidió retirar el medicamento.
Dos años después, ingresó por una cefalea asociada con una sensación vertiginosa precedida por síntomas respiratorios altos. Se inició metilprednisolona y se solicitó RMN de neuroeje, cuyo resultado no evidenció lesiones. Se solicitaron nuevamente bandas oligoclonales en LCR, y Ac anti-AQP4 y Ac anti-MOG en suero, para definir la continuidad del manejo modificador de la enfermedad. El citoquímico de LCR resultó normal; pero fue positivo el resultado de los Ac anti-MOG en suero. Se solicitó valoración por parte del servicio de inmunología e infectología, y se inició azatioprina, con mala adherencia referida posteriormente por el paciente.
Un año después, el paciente reingresó en contexto de cefalea occipital tipo presión, de intensidad fluctuante (con un promedio 4/10 constante), sin respuesta al acetaminofén; asociada a tinnitus, sin fotofobia, sin fonofobia, sin fosfenos, sin náuseas ni emesis. Adicionalmente, refirió dolor dorsolumbar, sensación vertiginosa y sensación subjetiva de debilidad, inicialmente en los miembros inferiores, y posteriormente, generalizada. Indicó un cuadro de síntomas respiratorios la semana anterior. Se solicitó una RMN de neuroeje simple y contrastada, con RMN cerebral sin lesiones pero con evidencia de realce leptomeníngeo. La RMN de columna fue normal.
Después se le realizó una punción lumbar, cuyo resultado fue: proteínas de 59 mg/dl, glucosa de 48,98 mg/dl con glucosa central de 108 mg/dl, leucocitos de 40 y diferencial con linfocitos de 20. Se consideró pleocitosis y proteínas en límite superior. Los exámenes Gram, cultivo, FilmArray, hidróxido de potasio y prueba de adenosina desaminasa fueron negativos. El paciente fue valorado por infectopediatría, que descartó alguna patología infecciosa. En este momento, el paciente venía en manejo con azatioprina, indicada a 1,58 mg/kg cada día. Presentó mejoría clínica y se consideró egreso con seguimiento estricto.
A los 10 días, el paciente reconsultó dado un cuadro de 3 días de evolución, consistente en diplopía horizontal, inestabilidad para la marcha con lateropulsión a la derecha, sensación vertiginosa y cefalea en la región frontal bilateral opresiva de intensidad leve-moderada. Se solicitó una nueva RMN simple y contrastada de neuroeje que evidenció lesiones hiperintensas con realce con el medio de contraste en los pedúnculos cerebelosos de forma bilateral y compromiso pontino (figura 5).
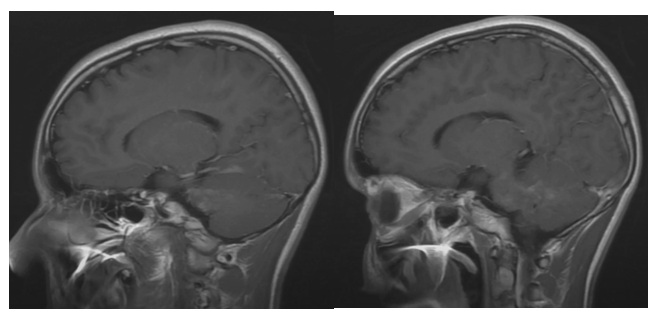
Dados los resultados de la RMN, se consideró recaída, se iniciaron pulsos de metilprednisolona y se continuó con prednisolona a 1 mg/kg cada día. Tuvo una mejoría clínica significativa de los síntomas a las 48-72 horas. Se planteó un cambio de manejo modificador de la enfermedad a Ig mensual, y con ayuda del servicio de psicología al paciente y sus familiares, se logró una adecuada concientización sobre el uso regular del medicamento. Se dio egreso, hasta el momento con adecuada evolución clínica, sin nuevas recaídas de la enfermedad.
Discusión
La enfermedad asociada con Ac anti-MOG (MOGAD) es una condición inflamatoria desmielinizante del SNC, caracterizada por un curso monofásico o recurrente de déficits neurológicos que no cumplen criterios para EM típica u otra enfermedad neuroinflamatoria conocida, especialmente NMOSD, y que ocurre en presencia de Ac anti-MOG positivos (7,12).
Respecto a la epidemiología, la enfermedad por anticuerpos anti-MOG se ha considerado una patología rara; sin embargo, tiene una incidencia mayor en pacientes pediátricos versus adultos (4,5). La frecuencia de anticuerpos anti-MOG positivos en el primer síndrome desmielinizante agudo en niños es del 40 % y del 24 % en síndromes recurrentes (8). No se han identificado diferencias según raza o etnia, y respecto al sexo, la frecuencia en pacientes pediátricos es igual, pero en la edad adulta existe una leve predominancia en el sexo femenino (8). No se ha asociado hasta la fecha con otras enfermedades autoinmunes, HLA o malignidad específica (8).
En el caso presentado, un adolescente inició sus síntomas a los 11 años de edad, con un cuadro de neuritis óptica con hallazgos clínicos e imagenológicos característicos de MOGAD, dados por el compromiso bilateral, que se presenta hasta en el 75 % de los casos; pérdida de agudeza visual severa al inicio de la enfermedad; edema de papila prominente; ausencia de compromiso del quiasma y del tracto óptico, y recuperación completa de los síntomas, al mostrar una buena respuesta a la terapia con corticoesteroides (3,4). También es más común en la MOGAD, en sus diagnósticos diferenciales principales (EM o NMOSD), una enfermedad prodrómica previa a los eventos, lo cual se evidenció en este caso.
Después, el paciente presentó un síndrome de tallo con compromiso cerebeloso, el cual se consideraba una condición altamente específica de NMOSD; sin embargo, se ha visto también en MOGAD, lo que constituye un fenotipo NMOSD-like. Por último, reingresó por una mala adherencia a la azatioprina y, nuevamente, con diagnóstico de síndrome de tallo y compromiso cerebeloso. Dado lo anterior, se considera un paciente con fenotipo NMOSD-like recurrente, un síndrome raro en la edad pediátrica (3).
En principio, los trastornos del espectro de la neuromielitis óptica se caracterizaban por episodios de neuritis óptica y mielitis transversa simultáneas o recurrentes; pero desde 2015 se amplió para incluir en el espectro síndromes de tallo y formas limitadas (5). Para apoyar su diagnóstico, se planteó la presencia de Ac AQP4, los cuales son raros en la edad pediátrica. Posteriormente, se comenzaron a identificar fenotipos similares en pacientes con estos anticuerpos negativos, y Ac anti-MOG positivos (7).
Dentro de otras diferencias entre las dos entidades, los pacientes con fenotipo NMOSD-like y Ac AQP4 positivos usualmente se asocian con otras enfermedades autoinmunes. El curso de la enfermedad es con mucha más frecuencia recurrente y existe un alto riesgo de pobre recuperación (5). También existen diferencias dentro de los fenotipos: en pacientes con neuritis óptica, Ac AQP4+ y Ac anti-MOG hay una afectación del quiasma y tracto óptico; en la mielitis transversa se comprometen regiones cervicotorácicas, no el cono medular, y es frecuente el síndrome de área postrema y el síndrome de tallo aislado (4,7).
También es relevante tener en cuenta que estudios recientes han mostrado la presencia de realce leptomeníngeo y pleocitosis en pacientes con MOGAD. De hecho, literatura principalmente de pacientes adultos indica que existe realce leptomeníngeo en el 2 %-15 % de pacientes con diagnóstico de MOGAD, pleocitosis en el 50 % (con predominio de linfocitos y monocitos) y casos de instauración de la enfermedad con meningitis aséptica, que luego puede progresar a alguno de los otros fenotipos descritos (3,8,15).
Lo más común es que una vez el paciente presenta los síntomas, en la neuroimagen se evidencien lesiones hiperintensas en T2 y realce leptomeníngeo (16). Sin embargo, Gombolay y Gadde (15) expusieron que 4 de cada 11 pacientes con Ac anti-MOG positivos tuvieron únicamente realce al inicio de la enfermedad y 3 de ellos desarrollaron lesiones desmielinizantes y síntomas después. De hecho, algunos autores consideran que los pacientes con realce leptomeníngeo en la neuroimagen tienen un mayor riesgo de una recaída, lo cual coincide con este caso, y expone la necesidad de ofrecer un seguimiento cercano en los pacientes en los que se encuentren estos hallazgos (14).
Respecto al manejo, un primer episodio desmielinizante asociado con Ac anti-MOG positivo no es indicativo de requerir tratamiento de mantenimiento, dado que la mayoría de los pacientes no presentan recurrencia y tienen buen pronóstico (1,17). Es un desafío identificar de forma temprana pacientes con alto riesgo de recurrencia y déficits a largo plazo, en quienes la inmunoterapia mejoraría los desenlaces (13). Actualmente no existen ensayos clínicos sobre cuándo instaurarla y se ha considerado que falta claridad sobre la historia natural de la enfermedad. Dado lo anterior y los riesgos de infección conocidos en relación con agentes inmunosupresores, la recomendación actual es iniciar el mantenimiento únicamente tras un segundo evento desmielinizante.
Algunas excepciones incluyen a pacientes con una pobre recuperación, 1-3 meses tras el evento inicial, y en quienes se da empeoramiento subclínico (por ejemplo, de neuritis óptica documentada en tomografía de coherencia óptica, o progresión asintomática de lesiones en RMN, aunque es muy raro en MOGAD, y en estos casos deberían considerarse otras patologías). También se debe individualizar su inicio, haciendo un balance de riesgo-beneficio, en pacientes con un largo intervalo de tiempo (>18 meses) entre el evento inicial y la primera recurrencia, que tienen buena recuperación tras el manejo agudo (13).
Respecto a las opciones de tratamiento, el micofenolato, la azatioprina, el rituximab y la Ig intravenosa mensual han mostrado reducir la tasa de recurrencia y estabilizar desenlaces (8,12). La opción terapéutica se debe basar en el perfil de efectos adversos del medicamento y las características del paciente, así como las preferencias de este último y sus padres (13). El acetato de glatiramer y el natalizumab han mostrado ser inefectivos en MOGAD, al igual que el β-interferón y el fingolimod (1,8).
Conclusiones
En pacientes pediátricos con síndromes desmielinizantes es importante solicitar la prueba de Ac anti-MOG y por método de cultivo celular, especialmente en pacientes con lesiones en el cerebro, o la médula, en la RMN, y con Ac anti-AQP4, en sangre y bandas oligoclonales, en LCR negativos, teniendo en cuenta la heterogeneidad de MOGAD en la edad pediátrica y su alta frecuencia de presentación en este grupo poblacional (5).
Se ha evidenciado que no existe una progresión gradual de la enfermedad, independiente de los eventos. Las secuelas son en su gran mayoría consecuencia directa del número de eventos y el fenotipo que presenta el paciente en cada caso (8,18); por esta razón, es de gran relevancia el diagnóstico temprano y un adecuado tratamiento modificador de la enfermedad para prevenir recurrencias y evitar secuelas, en aras de mejorar la calidad de vida de estos pacientes (3,12).
Referencias
1. Hur MH. Myelin oligodendrocyte glycoprotein antibody-associated disease: presentation, diagnosis, and management. Pediatr Ann. 2021;50(6):e254-8.
2. Di Pauli F, Reindl M, Berger T. New clinical implications of anti-myelin oligodendrocyte glycoprotein antibodies in children with CNS demyelinating diseases. Mult Scler Relat Disord. 2018;22(November 2017):35-7. https://doi.org/10.1016/j.msard.2018.02.023
3. Tenembaum SN. Pediatric demyelinating disease and anti-MOG antibody. Clin Exp Neuroimmunol. 2021;12(1):7-21. https://doi.org/10.1111/cen3.12627
4. Reindl M, Waters P. Myelin oligodendrocyte glycoprotein antibodies in neurological disease. Nat Rev Neurol. 2019;15(2):89-102. https://doi.org/10.1038/s41582-018-0112-x
5. Bruijstens AL, Lechner C, Flet-Berliac L, Deiva K, Neuteboom RF, Hemingway C, et al. E.U. paediatric MOG consortium consensus: Part 1 - Classification of clinical phenotypes of paediatric myelin oligodendrocyte glycoprotein antibody-associated disorders. Eur J Paediatr Neurol. 2020;29:2-13. https://doi.org/10.1016/j.ejpn.2020.10.006
6. Hemmer B, Archelos JJ, Hartung HP. New concepts in the immunopathogenesis of multiple sclerosis. Nat Rev Neurosci. 2002;3(4):291-301.
7. Alonso AS, Sakinis T, Pfeiffer HC, Sandvig I, Barlinn J, Marthinsen PB. Understanding pediatric neuro-immune disorder conflicts: a neuroradiologic approach in the molecular era. Radiographics. 2020;40(5):1395-411. https://doi.org/10.1148/rg.2020200032
8. Marignier R, Hacohen Y, Cobo-calvo A, Pröbstel A, Aktas O, Alexopoulos H, et al. Myelin-oligodendrocyte glycoprotein antibody-associated disease. Lancet Neurol. 2021 Sep;20(9):762-772. https://doi.org/10.1016/S1474-4422(21)00218-0
9. Banwell B, Bennett JL, Marignier R, Kim HJ, Brilot F, Flanagan EP, et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: International MOGAD Panel proposed criteria. Lancet Neurol. 2023 Mar;22(3):268-282. https://doi.org/10.1016/S1474-4422(22)00431-8
10. The North American Menopause Society. The 2017 hormone therapy position statement of the North American Menopause Society. Menopause. 2017;24(7):728-53. https://doi.org/10.1097/GME.0000000000001241
11. Armangue T, Capobianco M, de Chalus A, Laetitia G, Deiva K, Bruijstens AL, et al. E.U. paediatric MOG consortium consensus: Part 3 - Biomarkers of paediatric myelin oligodendrocyte glycoprotein antibody-associated disorders. Eur J Paediatr Neurol. 2020;29:22-31. https://doi.org/10.1016/j.ejpn.2020.11.001
12. Carnero Contentti E, Marrodan M, Correale J. Emerging drugs for the treatment of adult MOG-IgG-associated diseases. Expert Opin Emerg Drugs. 2021;26(2):75-8. https://doi.org/10.1080/14728214.2021.1919082
13. Bruijstens AL, Wendel EM, Lechner C, Bartels F, Finke C, Breu M, et al. E.U. paediatric MOG consortium consensus: Part 5 - Treatment of paediatric myelin oligodendrocyte glycoprotein antibody-associated disorders. Eur J Paediatr Neurol. 2020;29:41-53. https://doi.org/10.1016/j.ejpn.2020.10.005
14. Gadde JA, Wolf DS, Keller S, Gombolay GY. Rate of leptomeningeal enhancement in pediatric myelin oligodendrocyte glycoprotein antibody-associated encephalomyelitis. J Child Neurol. 2021;36(11):1042-6. https://doi.org/10.1177/08830738211025867
15. Gombolay GY, Gadde JA. Aseptic meningitis and leptomeningeal enhancement associated with anti-MOG antibodies: A review. J Neuroimmunol. 2021;358(April):577653. https://doi.org/10.1016/j.jneuroim.2021.577653
16. Baumann M, Bartels F, Finke C, Adamsbaum C. E. U. paediatric MOG consortium consensus: Part 2 e Neuroimaging features of paediatric myelin oligodendrocyte glycoprotein antibody-associated disorders. Eur J Paediatr Neurol. 2021;29(2020):0-6.
17. Chen JJ, Flanagan EP, Jitprapaikulsan J, López-Chiriboga A (Sebastian) S, Fryer JP, Leavitt JA, et al. Myelin oligodendrocyte glycoprotein antibody-positive optic neuritis: clinical characteristics, radiologic clues, and outcome. Am J Ophthalmol. 2018;195(12):8-15. https://doi.org/10.1016/j.ajo.2018.07.020
18. Bruijstens AL, Breu M, Wendel EM, Wassmer E, Lim M, Neuteboom RF, et al. E.U. paediatric MOG consortium consensus: Part 4 - Outcome of paediatric myelin oligodendrocyte glycoprotein antibody-associated disorders. Eur J Paediatr Neurol. 2020;29:32-40. https://doi.org/10.1016/j.ejpn.2020.10.007
Notas de autor
aAutora de correspondencia: est.isabella.lince@unimilitar.edu.co
Información adicional
Cómo citar: Lince-Rivera I, Martínez-Córdoba N,
Araújo Polanía AF. Fenotipo
NMOSD-like recurrente en un paciente pediátrico con enfermedad
por anticuerpos anti-MOG: presentación de caso. Salud. 2025;2. https://doi.org/10.11144/Javeriana.salud2.nmos
