





APA
ISO 690-2
Harvard
Haga clic en un formato de citación
Cortisol elevado como confusor en un caso de hiperplasia suprarrenal congénita por déficit de 21 hidroxilasa
Elevated Cortisol as a Confounder in a Case of Congenital Adrenal Hyperplasia due to 21-Hydroxylase Deficiency
Nydiacamila Suárez-Ahumada ![]() , Jairo Alejandro Obando
, Jairo Alejandro Obando ![]() , Isabella Lince-Rivera
, Isabella Lince-Rivera ![]() , Audrey Mary Matallana
, Audrey Mary Matallana ![]()
Cortisol elevado como confusor en un caso de hiperplasia suprarrenal congénita por déficit de 21 hidroxilasa
Salud Javeriana, vol. 1, 2024
Pontificia Universidad Javeriana
Nydiacamila Suárez-Ahumada a nydia.suarez@correounivalle.edu.co
Universidad del Valle, Colombia
Jairo Alejandro Obando
Universidad del Valle, Colombia
Isabella Lince-Rivera
Pontificia Universidad Javeriana, Colombia
Audrey Mary Matallana
Universidad del Valle, Colombia
Recibido: 12 marzo 2024
Aceptado: 21 agosto 2024
Resumen: La hiperplasia suprarrenal congénita (HSC) por déficit de 21 hidroxilasa (21-OH) es una enfermedad rara, caracterizada por una alteración en la producción de cortisol, aldosterona y andrógenos. El artículo presenta el caso de un bebé de dos meses de edad que ingresó al servicio de urgencias por desnutrición aguda severa, sin antecedentes relevantes, con hiperpigmentación de areolas y longitud de pene aumentada para la edad. En los exámenes paraclínicos se encontró hiponatremia severa, hipercalemia, hiperandorgenemia, corticotropina elevada, pero concentraciones elevadas de cortisol. Ante la sospecha clínica de HSC, a pesar del cortisol alto, se inició remplazo hormonal, y los estudios genéticos confirmaron la mutación en el gen CYP21A2, que condiciona el déficit de 21-OH. De este caso se resalta que una sospecha fuerte de HSC, por los hallazgos clínicos y el uso de pruebas genéticas, a pesar de los factores de confusión como niveles altos de cortisol, llevan a un adecuado diagnóstico y tratamiento. Adicionalmente, se hace hincapié en la importancia del tamizaje metabólico neonatal, para la detección temprana de una patología potencialmente mortal, que puede pasar inadvertida, y su tratamiento, que puede retrasarse y derivar en falla en el medro y crisis salina, como en este caso.
Palabras clave:hiperplasia adrenal congénita, cortisol, 21 hidroxilasa, pediatría.
Abstract: Congenital adrenal hyperplasia (CAH) due to 21-hydroxylase (21-OH) deficiency is a rare disease characterized by impaired cortisol, aldosterone and androgen production. We present the case of a 2-month-old male who was admitted to the emergency department for severe acute malnutrition, without relevant history, with hyperpigmentation of the areolae and increased penile length for his age. Tests showed severe hyponatremia, hyperkalemia, hyperandrogenemia, elevated ACTH, but elevated cortisol levels. Given the clinical suspicion of CAH despite the high cortisol levels, hormone replacement was started, and genetic studies confirmed the mutation in the CYP21A2 gene that causes 21-OH deficiency. This case highlights that a high suspicion of CAH by clinical findings and the use of genetic testing, despite confounding factors such as high cortisol levels, lead to an adequate diagnosis and treatment. Additionally, emphasis is placed on the importance of neonatal metabolic screening for early detection of a potentially fatal pathology that can go unnoticed, and its treatment can be delayed leading to failure to thrive and saline crisis as in this case.
Keywords: congenital adrenal hyperplasia, cortisol, 21 hydroxylase, pediatrics.
Introducción
La hiperplasia suprarrenal congénita (HSC) hace referencia a un espectro de trastornos autosómicos recesivos, caracterizados por una actividad disminuida de las enzimas involucradas en la esteroidogénesis (1). Por retroalimentación negativa, el déficit de cortisol aumenta la hormona adrenocorticotrópica (ACTH), que lleva a una hiperestimulación de la corteza suprarrenal, una hiperplasia de las glándulas suprarrenales y una elevación de los precursores previos al bloqueo enzimático (2).
El déficit de 21 hidroxilasa (21-OH) es la causa más común de HSC. En países de Europa y América del Norte, la incidencia es de 1:15000 nacidos vivos y en Latinoamérica oscila entre 1 en 9000 a 20000 nacidos vivos (3-5). Dependiendo de la actividad enzimática residual de la 21-OH, los pacientes presentan grados variables de deficiencia de glucocorticoides, exceso de andrógenos con o sin deficiencia de mineralocorticoides (2).
La forma tipo perdedora de sal es la variedad más grave, porque se reduce la formación de cortisol y aldosterona; mientras que la sobreproducción de precursores implica la producción de andrógenos (2). Esto conduce a una virilización excesiva en pacientes XY o a una virilización en pacientes XX; además de crisis perdedoras de sal, caracterizadas por hiponatremia, deshidratación, hiperpotasemia y acidosis metabólica secundarias al déficit del principal mineralocorticoide, pues la aldosterona regula la homeostasis del sodio, reteniéndolo a nivel tubular y arrastrando agua concomitantemente, lo cual mantiene la volemia (2).
La deficiencia de cortisol se asocia con una función cardiaca débil y pobre respuesta a las catecolaminas, así como con disminución de la tasa de filtración glomerular y aumento de la secreción de la hormona antidiurética (6). El perfil hormonal prototípico en esta forma es 17-OHP alta, ACTH, andrógenos altos y cortisol bajo (7). En la figura 1 se muestra la vía cascada metabólica adrenal normal y se compara con el déficit de 21-OH y los hallazgos del caso clínico.

Nota: con las flechas delgadas azules se describe la vía enzimática adrenal normal. En color negro se señalan las alteraciones de los metabolitos que se encuentran en el déficit de 21-OH, y el color verde se compara los hallazgos encontrados en el caso.
El diagnóstico se basa en la sospecha clínica y el perfil hormonal inicial. Es infrecuente encontrar concentraciones normales o elevadas de cortisol (8); sin embargo, en 2020 se informó de cortisol elevado en la sangre, debido a una reactividad cruzada de los anticuerpos inespecíficos utilizados en las pruebas de inmunoensayo. Ello puede retrasar el diagnóstico o hacer pensar en otros diagnósticos diferenciales, ante la clínica de virilización, hiponatremia, cortisol y ACTH elevados sin hipercortisolismo, como es el síndrome de resistencia primaria a los glucocorticoides (7). Este se caracteriza por una insensibilidad tisular a los glucocorticoides, causada por defectos genéticos en el gen NR3C1, que lleva a hipersecreción de la ACTH, así como a aumento de la producción de cortisol, andrógenos suprarrenales y precursores de esteroides con actividad mineralocorticoide (9).
La diferenciación requiere identificar la mutación específica a través de pruebas genéticas que deben estar ampliamente disponibles para la confirmación diagnóstica (8). La detección precoz de las formas clásicas de la HSC está incorporada en los programas de cribado neonatal, con ayuda de la medición de la 17-OHP en la muestra de sangre capilar obtenida del talón e impregnada en papel absorbente, extraída a las 48 horas de vida. Tiene como objetivos lograr la identificación presintomática de las formas clásicas severas y evitar la instauración de cuadros graves de deshidratación, shock y muerte, en especial en los recién nacidos varones con formas clásicas como en el caso que se describe (10).
A continuación, se presenta el caso de un paciente con HSC por déficit de 21-OH y concentraciones elevadas de cortisol, un hallazgo infrecuente.
Presentación del caso
El caso corresponde a un bebé de dos meses de edad, mestizo, procedente del área urbana, quien ingresó al servicio de urgencias, por pérdida de peso desde el nacimiento, sin otra sintomatología. Era alimentado con lactancia materna exclusiva a libre demanda. Dentro de los antecedentes perinatales se encontraban: nacimiento a término, con peso de 3890 gramos, longitud de 50,5 centímetros, sin que hubiera requerido hospitalización en el periodo neonatal.
Además, eran normales los valores de la hormona estimulante de la tiroides neonatal (3,3 mUI/L); sin antecedentes patológicos, y no había recibido ningún medicamento. No se informaron otros síntomas en la revisión por sistemas.
En el examen físico de ingreso presentó los siguientes signos vitales: frecuencia cardíaca de 140 latidos por minuto; frecuencia respiratoria de 30 revoluciones por minuto; saturación de oxígeno de un 95% con oxígeno ambiente; temperatura de 36,2°C, y tensión arterial de 86/60 mmHg. Por otra parte, su peso se encontraba en 3640 gramos, con una longitud de 53 centímetros. Peso/talla (P/T): −3,6 desviaciones estándar (DE), indicativo de una desnutrición aguda severa, sin signos de deshidratación, con hiperpigmentación en areolas y genitales, y longitud del pene aumentada para la edad (figura 2).

En el ingreso, los exámenes paraclínicos mostraron un trastorno hidroelectrolítico, por hiponatremia severa normovolémica de 106 Meq/L; hipercalemia de 6,2 Meq/L, y trastorno ácido base con acidosis metabólica (pH de 7,1; bicarbonato de 15,1) con brecha aniónica elevada (20) y glucemia basal de 107 mg/dl (tabla 1). A partir de estos resultados, se inició soporte y recuperación nutricional. Concomitantemente, se buscaron las causas de la desnutrición secundaria, como tubulopatías, alteraciones renales o fibrosis quística. Para ello, se documentaron como normales los electrolitos en la orina, la ecografía renal y los electrolitos en el sudor; por lo cual se descartaron (tabla 1).
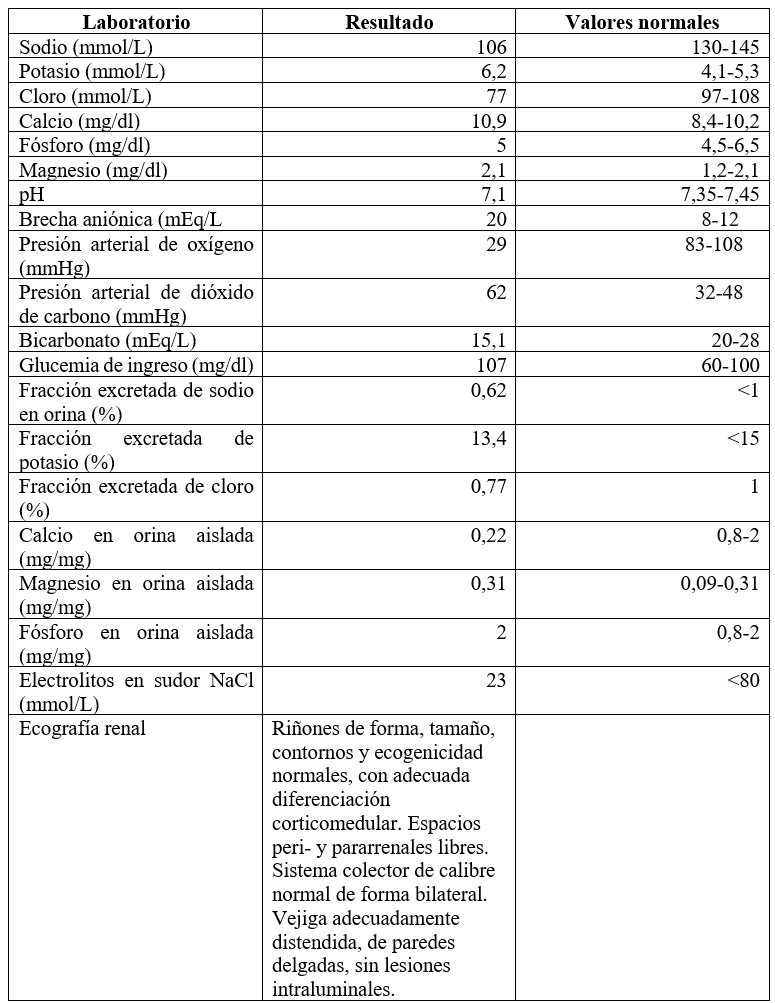
Por las alteraciones hidroelectrolíticas y virilización, se solicitó cariotipo con reporte 46 XY y un perfil hormonal. Este mostró concentraciones elevadas de la ACTH en la sangre (52,84 µg/dL; valores normales: 4,7-48,8 pg/mL); así como valores elevados de andrógenos, dehidroepiandrosterona sulfato (DHEAS) (274 µg/dL; valores normales: 31,6-214,1 µg/dL) y androstenediona (43,5 ng/ml; valores normales: <0,29 ng/mL).
En el contexto del paciente desnutrido, se solicitó testosterona libre, cuyo resultado fue de cantidades altas (5,4 pg/mL; valores normales: <1,80 pg/mL). La aldosterona fue normal (6,44 ng/dL; valores de referencia: 2,52-39,2 ng/dL); mientras que las concentraciones de cortisol fueron elevadas (61,6 µg/dl; valores normales: 4,46-22,7 µg/dL). No se realizaron exámenes para medir los niveles de 17-OH progesterona, por falta de disponibilidad en la institución (tabla 2).

Ante los hallazgos de virilización, hiponatremia y cortisol elevado sin hipercortisolismo, se consideró la posibilidad diagnóstica de síndrome de resistencia primaria al cortisol. Por ello, se indicó una prueba de supresión con dexametasona, y así disminuyeron las concentraciones de cortisol, pero no se suprimieron, lo cual apoyaba la sospecha diagnóstica de resistencia al cortisol. En consecuencia, y con el fin de generar la supresión en las concentraciones séricas de glucocorticoides, se inició manejo con dexametasona a dosis altas, con controles de cortisol en la sangre elevados; sin embargo, persistían la hiponatremia, la hipercalemia severa y la poliuria, a pesar de las medidas correctivas.
Por la clínica del paciente y la persistencia de las alteraciones hidroelectrolíticas, a pesar de la singularidad de los resultados del cortisol, no se descartó la sospecha diagnostica de HSC; de ahí que se haya solicitado una prueba genética con exoma, dirigida a las alteraciones del cortisol. De acuerdo con sus resultados, había una variante patogénica en hemicigosis/homocigosis en el gen CYP21A2c 293-13C/A>G, que corroboró el diagnóstico de HSC por déficit de 21-OH. Ante ello, se cambió el manejo de dexametasona por hidrocortisona y se inició suplencia hormonal con fludrocortisona. De este modo, se logró la normalización de los electrolitos y de los parámetros hormonales en la sangre. Durante los controles ambulatorios, el paciente mantuvo la suplementación hormonal con prednisolona, por la disponibilidad oral, y con fludrocortisona, con adecuada evolución.
Discusión
La HSC por déficit de 21-OH es la enfermedad congénita más común y representa del 90% al 95% de todos los casos; además, está asociada con mutaciones en el gen CYP21A2, como se describe en este caso (11). Dependiendo de la severidad del déficit enzimático, existen varios fenotipos. Su clasificación clínica se basa en el tipo de manifestaciones y la edad de presentación; por lo general, se divide en dos grandes grupos: clásica y no clásica (12). La HSC clásica se subdivide en dos formas: HSC clásica perdedora de sal (grave) e HSC clásica virilizante simple.
La primera se caracteriza por crisis salinas y trastornos de diferenciación sexual, como el caso descrito, y la forma virilizante simple, por signos de virilización más sutiles y la ausencia de crisis salina, con un diagnóstico más tardío, porque es menos frecuente (13). En la forma clásica perdedora de sal concurren el hiperandrogenismo, concentraciones bajas de cortisol y déficit completo de aldosterona. Se manifiesta con deshidratación, shock, hiponatremia, hipercalemia y acidosis metabólica, y es potencialmente mortal en las primeras semanas de vida (6).
En el caso presentado, a pesar de las alteraciones metabólicas marcadas y de corresponder con una forma clásica perdedora de sal, la única manifestación fue la falla en el medro, sin inestabilidad clínica. Aparte de lo anterior, concuerda con las características clínicas que se han descrito en cohortes de pacientes pediátricos con HSC en Colombia, como la mediana de edad al diagnóstico de dos meses y donde el subtipo más frecuente es la HSC clásica perdedora de sal, con trastorno de diferenciación sexual y crisis salina (14).
Es habitual que los varones con HSC por déficit de 21-OH se diagnostiquen de forma más tardía y no en la etapa neonatal en países sin protocolos de tamizaje neonatal, puesto que no presentan ambigüedad sexual, como su contraparte femenina, que alerta la posibilidad etiológica (15), como en este caso. Por ello, el tamizaje neonatal reduce el tiempo del diagnóstico de la HSC clásica perdedora de sal y permite un tratamiento presintomático que evita sus complicaciones (16)
En ausencia de tamizaje metabólico, el diagnóstico se logra sobre la base de la historia clínica y las pruebas bioquímicas y hormonales relevantes (7). Los resultados de los estudios hormonales se caracterizan por niveles séricos marcadamente elevados del precursor 17-OHP y se podrían confirmar con la prueba de estimulación con ACTH midiendo los niveles basales y estimulados de 17-OHP (8), en los casos que se requiera, sin embargo su medición no está ampliamente disponible en países de bajos y medianos ingresos. Otros estudios de laboratorio que orientan el diagnóstico son niveles de cortisol y aldosterona bajos con niveles elevados de progesterona, androstenediona, testosterona, DHEAS, ATCH (6-8). En este caso a pesar de tener valores elevados de andrógenos y ACTH, los de cortisol fueron elevados.
Agrawal et al. (7) también informaron un caso de HSC clásica perdedora de sal por déficit de 21-OH, con valores altos de cortisol en una paciente de 45 días de nacida, con crisis salina y genitales ambiguos. En esa revisión se describen las posibilidades diagnósticas ante estos hallazgos, como deficiencia de 21-OH con uso previo de hidrocortisona. Ello se descartó en nuestro paciente, a partir de la historia clínica, deficiencia de 21-OH y globulina transportadora de cortisol alta coexistente, la cual tampoco correspondía a nuestro caso y síndrome de resistencia primaria a glucocorticoides (7).
Este último se consideró como diagnóstico diferencial, ante los hallazgos de virilización, hiponatremia, ACTH y cortisol elevado sin signos de hipercortisolismo; por lo tanto, se suprimió la dexametasona y así disminuyeron las concentraciones de cortisol; pero no se suprimieron. Ello apoyaba la sospecha diagnóstica de resistencia al cortisol, manejada con dosis altas de dexametasona (9). A pesar del tratamiento, en este caso, persistieron las alteraciones hidroelectrolíticas y, por ese motivo, no se descartó la HSC.
Los hallazgos paraclínicos inusuales, como cortisol elevado, pueden ser un factor confusor en el abordaje diagnóstico de la HSC por déficit de 21-OH, como lo describen Agrawal et al. (7). El inmunoensayo quimioluminiscente, comúnmente utilizado para la medición de cortisol, es propenso a una variabilidad interensayo clínicamente significativa (7). Esto ocurre debido a la naturaleza inespecífica de los anticuerpos anticortisol usados, que tienen reactividad cruzada con precursores de cortisol estructuralmente similares, como 17-OHP, 11-desoxicortisol y 21-desoxicortisol, en pacientes con deficiencia de 21-OH, en quienes están significativamente elevados la 17-OHP y la 21-deoxicortisol (7).
En nuestro caso, el cortisol se midió con una técnica de inmunoensayo competitivo, que se basa en la competencia entre el cortisol presente en la muestra y un conjugado de cortisol sobre un anticuerpo biotinilado. El complejo antígeno-anticuerpo es capturado por la estreptavidina y se mide con una reacción luminiscente. En este caso, la reactividad cruzada informada por el fabricante es del 1,15%, lo cual podría explicar los valores elevados de cortisol.
La cromatografía líquida-espectrometría de masas en tándem es el método estándar para medir el cortisol, pero es costosa y no está disponible en todas las instituciones (7). De ahí que la genotipificación se esté convirtiendo progresivamente en el estudio fundamental de estos pacientes, en especial para confirmar casos difíciles. Un análisis correcto de los casos de deficiencia de 21-OH implica la secuenciación completa de todo el gen versus el análisis dirigido de variantes patogénicas, ya que permite detectar variantes patógenas no detectadas por el análisis dirigido y también es capaz de detectar nuevas variantes.
Los métodos de detección de mutaciones basados en la reacción en cadena de la polimerasa con secuenciación del gen completo y amplificación de la sonda dependiente de ligadura múltiple son los exámenes de referencia para genotipificar el gen CYP21A2 (1). La tasa de detección de variantes patogénicas del gen CYP21A2, mediante secuenciación genética, es aproximadamente del 95% al 98% en pacientes con deficiencia de 21-OH (17).
En este caso, se solicitó un exoma dirigido a genes relacionados con alteraciones del cortisol, que detectó una variante patogénica en hemicigosis/homocigosis en el gen CYP21A2c.293-13C/A>G, que informó el diagnóstico de déficit de 21-OH. Esta variante está presente en aproximadamente el 20-30% de los alelos mutados en pacientes con HSC (17). Se caracteriza por la sustitución del nucleótido adenosina o citosina por guanina, que causa un empalme aberrante del intrón 2 con retención de 19 nucleótidos normalmente empalmados del ARNm. Esto resulta en un cambio en el marco de lectura traduccional y en la alteración de la función de la enzima 21-OH (1).
Conclusión
Del caso presentado se destaca que las concentraciones elevadas de cortisol pueden actuar como un factor confusor en el diagnóstico de HSC por déficit de 21-OH y que, en algunos escenarios, puede haber limitaciones para un diagnóstico correcto inicial.
Es preciso conocer y estar familiarizados con este tipo de factores, que pueden llevar a estudios y tratamientos innecesarios. Adicionalmente, debe predominar la sospecha clínica, de acuerdo con las manifestaciones más frecuentes, como hiperandrogenismo, trastorno de la diferenciación sexual y crisis salina.
También se hace hincapié en la importancia de contar con pruebas genéticas/moleculares para la confirmación diagnóstica temprana; así como en la importancia de la implementación activa de la tamización neonatal de esta patología, a fin de disminuir los retrasos en el diagnóstico y el tratamiento, y reducir, de este modo, tiempos de hospitalización, morbilidad y mortalidad.
Consideraciones éticas
Se obtuvo el consentimiento informado por escrito de parte de los padres del paciente y fue avalado por el Comité de Ética del Hospital Universitario del Valle Evaristo García, para la publicación de este informe de caso y las imágenes que lo acompañan.
Conflicto de intereses
Los autores no declaran conflictos de intereses.
Referencias
1. Pignatelli D, Carvalho BL, Palmeiro A, Barros A, Guerreiro SG, Maçut D. The complexities in genotyping of congenital adrenal hyperplasia: 21-hydroxylase deficiency. Front Endocrinol (Lausanne). 2019;10:432. https://doi.org/10.3389/fendo.2020.00113
2. Merke DP, Auchus RJ. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. N Engl J Med. 2020 Sep 24;383(13):1248-61. https://doi.org/10.1056/NEJMra1909786
3. Van der Kamp HJ, Wit JM. Neonatal screening for congenital adrenal hyperplasia. Eur J Endocrinol. 2004 Nov;151 Suppl 3(3). https://doi.org/10.1530/eje.0.151u071
4. Pezzuti IL, Barra CB, Mantovani RM, Januário JN, Silva IN. A three-year follow-up of congenital adrenal hyperplasia newborn screening. J Pediatr (Rio J). 2014;90(3):300-7. https://doi.org/10.1016/j.jped.2013.09.007
5. Gruñeiro-Papendieck L, Chiesa A, Mendez V, Prieto L. Neonatal screening for congenital adrenal hyperplasia: experience and results in Argentina. J Pediatr Endocrinol Metab. 2008; 21(1):73-8. https://doi.org/10.1515/jpem.2008.21.1.73
6. Speiser PW, White PC. Congenital adrenal hyperplasia. N Engl J Med. 2003 Aug 21;349(8):776-88. https://doi.org/10.1056/NEJMra021561
7. Agrawal N, Chakraborty PP, Sinha A, Maiti A. False elevation of serum cortisol in chemiluminescence immunoassay by Siemens Advia Centaur XP system in 21-hydroxylase deficiency: an “endocrine laboma”. BMJ Case Rep. 2020;13(9):e235450. https://doi.org/10.1136/bcr-2020-235450
8. Witchel SF. Congenital adrenal hyperplasia. J Pediatr Adolesc Gynecol. 2017;30(5):520-34. https://doi.org/10.1016/j.jpag.2017.04.001
9. Nicolaides NC, Charmandari E. Chrousos syndrome: from molecular pathogenesis to therapeutic management. Eur J Clin Invest. 2015;45(5):504-14. https://doi.org/10.1111/eci.12426
10. Dulín Iñiguez E, Ezquieta Zubicaray B. Newborn screening of congenital adrenal hyperplasia. Endocrinol Diabetes Nutr. 2018;65(1):1-4. https://doi.org/10.1016/j.endinu.2017.11.001
11. Riepe FG, Sippell WG. Recent advances in diagnosis, treatment, and outcome of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Rev Endocr Metab Disord. 2007;8(4):349-63. https://doi.org/10.1007/s11154-007-9053-1
12. Carmina E, Dewailly D, Escobar-Morreale HF, Kelestimur F, Moran C, Oberfield S, et al. Non-classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency revisited: an update with a special focus on adolescent and adult women. Hum Reprod Update. 2017;23(5):580-99. https://doi.org/10.1093/humupd/dmx014
13. Auchus RJ, Witchel SF, Leight KR, Aisenberg J, Azziz R, Bachega TA, et al. Guidelines for the Development of Comprehensive Care Centers for Congenital Adrenal Hyperplasia: guidance from the CARES Foundation Initiative. Int J Pediatr Endocrinol. 2010;1-17. https://doi.org/10.1155/2010/275213
14. Suárez DV, Matorel E, Niño-Serna L, Toro-Ramos M. Caracterización de una cohorte de pacientes pediátricos con hiperplasia suprarrenal congénita. Andes Pediátr. 2022;93(4):511-9. https://doi.org/10.32641/andespediatr.v93i4.4003
15. Van der Grinten HLC, Speiser PW, Faisal Ahmed S, Arlt W, Auchus RJ, Falhammar H, et al. Congenital adrenal hyperplasia-current insights in pathophysiology, diagnostics, and management. Endocr Rev. 2022;43(1):91-159. https://doi.org/10.1210/endrev/bnab016
16. Therrell BL, Berenbaum SA, Manter-Kapanke V, Simmank J, Korman K, Prentice L, et al. Results of screening 1.9 million Texas newborns for 21-hydroxylase- deficient congenital adrenal hyperplasia. Pediatrics. 1998 Apr;101(4 I):583-90. https://doi.org/10.1542/peds.101.4.583
17. Li H, Zhu X, Yang Y, Wang W, Mao A, Li J, et al. Long-read sequencing: an effective method for genetic analysis of CYP21A2 variation in congenital adrenal hyperplasia. Clin Chim Acta. 2023 Jul 1;547:117419. https://doi.org/10.1016/j.cca.2023.117419
Notas de autor
aAutora de correspondencia: nydia.suarez@correounivalle.edu.co
Información adicional
Cómo citar: Suárez-Ahumada N, Obando JA, Lince-Rivera I, Matallana AM. Cortisol elevado como confusor en un caso de hiperplasia suprarrenal congénita por déficit de 21 hidroxilasa. Salud. 2024;1. https://doi.org/10.11144/Javeriana.salud1.csfc
